Tutorials
Transcriptome reference generation
The first steps of cerberus are to generate a reference set of transcript start sites (TSSs), intron chains (ICs), and transcript end sites (TESs). TSSs and TESs can either be added from GTFs or BED files. ICs can be added from GTFs.
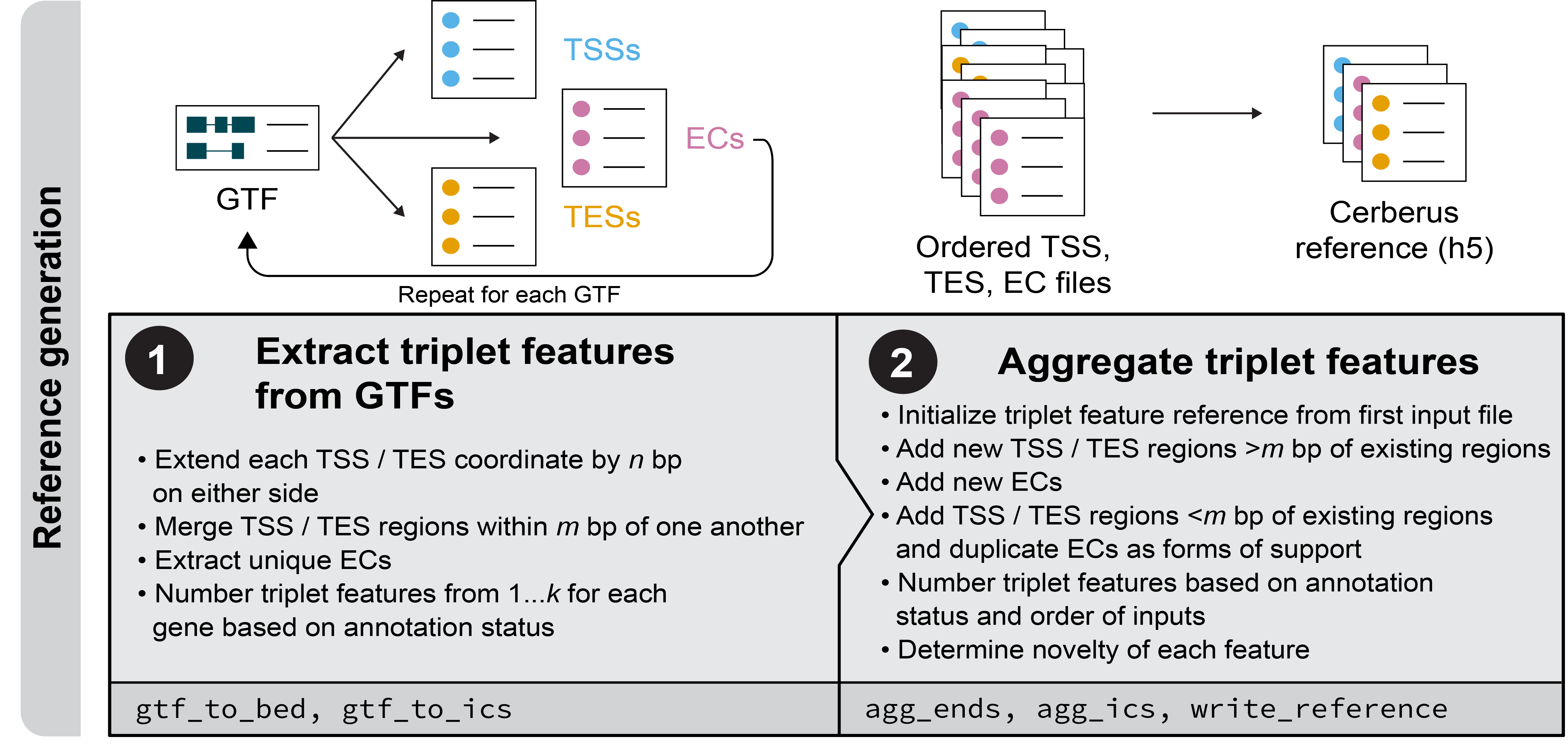
Extracting triplet features from GTFs
To generate BED files for the TSSs and TESs from a GTF file, use cerberus gtf_to_bed.
Usage: cerberus gtf_to_bed [OPTIONS]
Options:
--gtf TEXT GTF file [required]
--mode TEXT Choose tss or tes [required]
-o TEXT Output file name [required]
--dist INTEGER Distance (bp) to extend regions on either side [default:
50]
--slack INTEGER Distance allowable for merging regions [default: 50]
--help Show this message and exit.
Output file format:
Chromosome |
Start |
Stop |
Name |
Score |
Strand |
ThickStart |
|---|---|---|---|---|---|---|
chr1 |
169804296 |
169804436 |
ENSG00000000460_2 |
. |
cerberus |
|
chr1 |
169807786 |
169807887 |
ENSG00000000460_4 |
. |
cerberus |
The
ThickStartcolumn is used to indicate the source of the data
To generate tab-separated files for the ICs from a GTF file, use cerberus gtf_to_ics.
Usage: cerberus gtf_to_ics [OPTIONS]
Options:
--gtf TEXT GTF file [required]
-o TEXT Output file name [required]
--help Show this message and exit.
Output file format:
Chromosome |
Strand |
Coordinates |
Name |
|---|---|---|---|
chr1 |
ENSG00000000460_1 |
||
chr1 |
54-87 |
ENSG00000000460_2 |
Coordinates is a
--separated list of the internal splice sites usedMonoexonic transcripts are assigned the coordinate
-
Aggregating TSSs and TESs across multiple sources
Cerberus can aggregate information to support the existence or creation of TSS or TES
regions across multiple bed files. To perform this aggregation, use cerberus agg_ends.
Usage: cerberus agg_ends [OPTIONS]
Options:
--input TEXT Path to config file. Each line contains file path, whether
to add ends (True / False), whether to use as a reference,
source name [required]
--mode TEXT Choose tss or tes [required]
--slack INTEGER Distance (bp) allowable for merging regions [default: 20]
-o TEXT Output file name [required]
--help Show this message and exit.
Notes on behavior:
TSSs / TESs will be numbered from 1…n for each gene based on
MANE status, appris_principal status, and basic_set status of TSS / TES (if tags are present in GTF used to call regions)
Order of BED files in config file (earlier BED files will yield lower numbers)
BED files without gene IDs cannot be used to initialize new TSS / TES regions
After a region is initialized, the boundaries are fixed. Regions that are considered thereafter are simply added as additional forms of support for the already-initialized regions. The reasoning behind this is to avoid “super regions” that grow as new data is added.
Input BED file format:
Generally, BED files follow the BED file specifications
If you have gene ids in your BED file, they must be in the
namefield (4th column), and must be_separated with additional text that makes the name unique. Additionally, thethickStart(7th) column must contain the stringcerberusto indicate to Cerberus how to parse the gene ids. This is how the files output fromcerberus gtf_to_bedare automatically formatted.
Chromosome |
Start |
Stop |
Name |
Score |
Strand |
ThickStart |
|---|---|---|---|---|---|---|
chr1 |
169804296 |
169804436 |
ENSG00000000460_2 |
. |
cerberus |
|
chr1 |
169807786 |
169807887 |
ENSG00000000460_4 |
. |
cerberus |
The
ThickStartcolumn is used to indicate the source of the data
Input config file format:
Comma-separated
No header
First BED must have
Trueadd ends settingBED files with
Trueadd ends setting must have gene ids and strandRecommended to use
Trueadd ends setting for GTFs that you are planning to annotate
BED file path |
Reference |
Add ends |
Source name |
|---|---|---|---|
path/to/bed/v40_tss.bed |
True |
True |
v40 |
path/to/bed/encode_tss.bed |
False |
True |
encode |
Output file format:
Chromosome |
Start |
Stop |
Name |
Score |
Strand |
Sources |
Novelty |
|---|---|---|---|---|---|---|---|
chr1 |
169794989 |
169795129 |
ENSG00000000460_1 |
. |
v40,v29,encode |
Known |
|
chr1 |
300 |
400 |
ENSG00000000460_2 |
. |
encode |
Novel |
The
ThickStartcolumn is used to indicate the sources, defined in the config file, for each region. Sources are comma-separated if there was more than one form of support for the region.The
ThickEndcolumn is used to indicate the novelty of the region. Regions not supported by one of the sources used as references will beNovel, others will beKnown.
Aggregating ICs across multiple sources
Cerberus can aggregate information to support the existence or creation of intron chains
across multiple datasets. To perform this aggregation, use cerberus agg_ics.
Usage: cerberus agg_ics [OPTIONS]
Options:
--input TEXT Path to config file. Each line contains file path, whether to
use as a reference, source name [required]
-o TEXT Output file name [required]
--help Show this message and exit.
Notes on behavior:
ICs will be numbered from 1…n for each gene based on
MANE status, appris_principal status, and basic_set status of IC (if tags are present in GTF used to call intron chains)
Order of IC TSV files in config file (earlier TSV files will yield lower numbers)
Input IC file format:
Output from
cerberus gtf_to_ics.
Chromosome |
Strand |
Coordinates |
Name |
|---|---|---|---|
chr1 |
ENSG00000000460_1 |
||
chr1 |
54-87 |
ENSG00000000460_2 |
Coordinates is a
--separated list of the internal splice sites usedMonoexonic transcripts are assigned the coordinate
-
Input config file format:
Comma-separated
No header
BED files with
Trueadd ends setting must have gene ids and strandRecommended to use
Trueadd ends setting for GTFs that you are planning to annotate
Output file format:
Chromosome |
Strand |
Coordinates |
Name |
source |
novelty |
|---|---|---|---|---|---|
chr1 |
ENSG00000000460_1 |
v40,v29 |
Known |
||
chr1 |
54-87 |
ENSG00000000460_2 |
encode |
NIC |
Coordinates is a
--separated list of the internal splice sites usedMonoexonic transcripts are assigned the coordinate
-Sources are comma-separated if there was more than one form of support for the intron chain.
Novelty of the intron chain is determined with respect to the sources used as references. Those supported are
Known. Others are assigned a SQANTI novelty category (ISM, NIC, NNC, and Unspliced).
Generating a cerberus reference in h5 format
The final step of reference generation is to write an h5 representation of the TSSs, ICs, and TESs present in the input data as a series of tables in h5 format.
Usage: cerberus write_reference [OPTIONS]
Options:
--tss TEXT TSS bed file output from `agg_ends` [required]
--tes TEXT TES bed file output from `agg_ends` [required]
--ics TEXT IC tsv file output from `agg_ics` [required]
-o TEXT Output .h5 file name
--help Show this message and exit.
Input TSS / TES file format:
Output from
cerberus agg_ends.
Chromosome |
Start |
Stop |
Name |
Score |
Strand |
Sources |
Novelty |
|---|---|---|---|---|---|---|---|
chr1 |
169794989 |
169795129 |
ENSG00000000460_1 |
. |
v40,v29,encode |
Known |
|
chr1 |
300 |
400 |
ENSG00000000460_2 |
. |
encode |
Novel |
The
ThickStartcolumn is used to indicate the sources, defined in the config file, for each region. Sources are comma-separated if there was more than one form of support for the region.The
ThickEndcolumn is used to indicate the novelty of the region. Regions not supported by one of the sources used as references will beNovel, others will beKnown.
Input IC file format:
Output from
cerberus agg_ics.
Chromosome |
Strand |
Coordinates |
Name |
source |
novelty |
|---|---|---|---|---|---|
chr1 |
ENSG00000000460_1 |
v40,v29 |
Known |
||
chr1 |
54-87 |
ENSG00000000460_2 |
encode |
NIC |
Coordinates is a
--separated list of the internal splice sites usedMonoexonic transcripts are assigned the coordinate
-Sources are comma-separated if there was more than one form of support for the intron chain.
Novelty of the intron chain is determined with respect to the sources used as references. Those supported are
Known. Others are assigned a SQANTI novelty category (ISM, NIC, NNC, and Unspliced).
Output cerberus reference h5 format:
CerberusAnnotation()object saved in h5 format.Can be read in in Python using
import cerberus
ca = cerberus.read(<ref name>)
TSS / TES regions accessible using:
ca.tss
ca.tes
Chromosome |
Start |
End |
Strand |
Name |
novelty |
source |
gene_id |
tss/tes |
|---|---|---|---|---|---|---|---|---|
chr1 |
169794989 |
169795129 |
ENSG00000000460_1 |
v40,v29 |
Known |
ENSG00000000460 |
1 |
|
chr1 |
169795358 |
169795459 |
ENSG00000000460_2 |
encode |
Novel |
ENSG00000000460 |
2 |
BED-inspired with a few added columns
Because end matching is less precise than intron chain matching, the Cerberus reference h5 also store the mapping between each input TSS / TES and what region in the reference (ie from ca.tss or ca.tes), if any, the input region matched too.
These maps are stored in:
ca.tss_map
ca.tes_map
Chromosome |
Start |
End |
Strand |
source |
Name |
|---|---|---|---|---|---|
chr1 |
169794989 |
169795129 |
v40 |
ENSG00000000460_1 |
|
chr1 |
169795358 |
169795459 |
encode |
ENSG00000000460_2 |
Each BED entry from BED files input to
cerberus agg_endswill have an entry here.Original coordinates are reported in the
Chromosome,Start,End, andStrandcolumns.Sourcecolumn has source name as defined incerberus agg_endsNamecolumn includes the final ID for the Cerberus end region that is in the TSS or TES table.If the region was not included in the final reference, the
Namecolumn will beNaN.
Intron chains accessible using:
ca.ic
Chromosome |
Strand |
Coordinates |
Name |
source |
novelty |
gene_id |
ic |
|---|---|---|---|---|---|---|---|
chr1 |
ENSG00000000460_1 |
v40,v29 |
Known |
ENSG0000000046 |
1 |
||
chr1 |
54-87 |
ENSG00000000460_2 |
encode |
NIC |
ENSG0000000046 |
2 |
Transcriptome annnotation
After generating an h5 Cerverys reference of TSSs, ICs, and TESs, you can now annotate any transcriptome in GTF format using the features present in your Cerberus reference; generating transcript triplets for each transcript in your transcriptome. Multiple transcriptomes can be annotated using the same Cerberus reference. You can then use this mapping to add the Cerberus transcript triplets to preexisting GTF files and abundance matrices.

Assigning transcript triplets to a transcriptome
Assign each transcript in a GTF file a Cerberus TSS, IC, and TES. You can annotate multiple transcriptomes using the same Cerberus reference.
Note: It is recommended that for each transcriptome you annotate, that you have extracted TSSs, ICs, and TESs from the transcriptome’s GTF and added them to the Cerberus reference. Otherwise you run the risk of encountering triplet features that are unannotated in the Cerberus reference.
cerberus annotate_transcriptome \
--gtf {gtf} \
--h5 {cerberus ref} \
--source {source name} \
-o {cerberus annot}
Output file format:
Update counts matrix with Cerberus IDs
Replace transcript IDs from your transcriptome in an abundance matrix with cerberus isoform triplets.
cerberus replace_ab_ids \
--h5 {annot} \
--ab {ab} \
--source {source} \
--collapse \
-o {update_ab}
Notes on behavior:
Some transcripts from a transcriptome may be assigned the same triplet. Using the
--collapseflag will deduplicate these in the abundance file and sum up the counts across each transcript with the same isoform triplet.
Input abundance file format:
Currently cerberus only works with TALON abundance matrices, but we want to support other formats in the future.
Update GTF with Cerberus IDs
Replace transcript IDs from your transcriptome in a GTF with cerberus isoform triplets.
cerberus replace_gtf_ids \
--h5 {annot} \
--gtf {gtf} \
--source {source} \
--update_ends \
--collapse \
-o {update_gtf}
Notes on behavior:
Some transcripts from a transcriptome may be assigned the same triplet. Using the
--collapseflag will deduplicate these in the abundance file and sum up the counts across each transcript with the same isoform triplet.Because cerberus uses regions to represent TSSs and TESs rather than single-bp coordinates, the ends of each of your transcripts are not representable in GTF format. We circumvent this by choosing the furthest upstream boundary of the region for the TSS and the furthest downstream boundary for the TES to report in the GTF when the
--update_endsoption is used. If it is not, the original ends from your transcriptome will be used.
Calculating gene triplets

After you’ve generated your final Cerberus annotation h5 file for as many
transcriptomes as you wish with cerberus annotate_transcriptome, you can now start to
calculate gene triplets as a metric of measuring transcriptional diversity.
There are a few different ways of doing this.
Calculating gene triplets from Cerberus sources
The most simple gene triplet is calculated from all of the isoforms present in a given transcriptome source. You can calculate and add these triplets to your CerberusAnnotation using the cerberus.get_source_triplets() function.
In Python, import cerberus, read in your CerberusAnnotation (output from cerberus annotate_transcriptome), compute source triplets, and add them to your CerberusAnnotation:
import cerberus
ca = cerberus.read('cerberus_annot.h5')
df = ca.get_source_triplets()
ca.add_triplets(df)
After adding these source triplets, you can save your updated CerberusAnnotation in h5 format. The triplets will be saved in the ca.triplets DataFrame slot.
ca.write('cerberus_annot_triplets.h5')
Calculating gene triplets from a list of transcriptomes
You may want to filter the isoforms used to calculate your gene triplets. For example, maybe you want to only consider isoforms that are expressed above a certain TPM level. To compute gene triplets from each gene from any arbitrary list of isoform triplets, use cerberus.get_subset_triplets():
import cerberus
ca = cerberus.read('cerberus_annot.h5')
source_name = 'highly_expressed'
tids = ['Gene[1,1,1]', 'Gene[1,2,1]']
df = ca.get_subset_triplets(tids, source_name)
ca.add_triplets(df)
Calculating gene triplets based on transcripts expressed in a sample
You also may want to compute gene triplets based on a the set of isoforms expressed in a given sample. For this analysis, we use Swan to store our sample metadata and expression information:
import swan_vis as swan
sg = swan.SwanGraph()
sg.add_annotation(input.annot)
sg.add_transcriptome(input.gtf, include_isms=True)
sg.add_abundance(input.ab)
sg.add_abundance(input.gene_ab, how='gene')
sg.add_metadata(input.meta)
sg.save_graph(output.prefix)
We can use the expression values and metadata stored in the SwanGraph to determine which isoforms are expressed in each sample, and use cerberus.get_expressed_triplets() to calculate these values:
import swan_vis as swan
import cerberus
sg = swan.read(swan_file)
ca = cerberus.read('cerberus_annot.h5')
source_name = 'sample_isos'
metadata_column = 'sample'
min_tpm = 1
df = ca.get_expressed_triplets(sg,
obs_col=metadata_column,
min_tpm=min_tpm,
source=source_name)
ca.add_triplets(df)
Calculating gene triplets directly from a GTF
In cases where you already have a finalized transcriptome and don’t wish to use the Cerberus tools to generate your transcriptome, you have the option to generate summary gene triplets directly from a GTF using a command line tool.
cerberus gtf_to_triplets
Usage: cerberus gtf_to_triplets [OPTIONS]
Options:
--gtf TEXT GTF file to replace ids in [required]
--source TEXT name of source in cerberus object to map from
[required]
--gene_id_col TEXT Attribute name in GTF w/ gene id
--gene_name_col TEXT Attribute name in GTF w/ gene name
-o TEXT Output file name [required]
--help Show this message and exit.
The output file will be a CerberusAnnotation h5 object that is directly compatible with Cereberus’ downstream plotting functions.
Calculating gene triplets from custom subset files
In the cases where the above gene triplet calculation functions don’t fulfill your exact needs, cerberus supports adding triplets for individual samples using a tab-separated isoform subset file. Coming soon